Aspects of the Biochemical Pharmacology of Psychotropic Drugs
By Daniel X. Freedman, 1966

Only a decade ago, psychopharmacology faced the future equipped more with prescient hopes than with substantive findings
[1]. Drugs were promised as tools to reveal the coding by which neurochemical as well as neurobehavioral sequences were regulated. Endogenous systems related to stress were to be unmasked and their relationship to behavior pathology revealed.
Therapeutic drugs were to be discovered which — if they did not specifically reverse behavior disorder— would at the least set into motion compensatory and inhibitory processes. Drugs thereby would directly or indirectly permit a more successful operation of those contingencies normally regulating behavior and keeping it within acceptable bounds.
Today, it is the expanding range of specific information which is perhaps overwhelming. Where neurophysiologists have been able sufficiently to map the intricate sequences of peripheral and central signals which comprise the controls for basic drive behaviors such as eating and drinking, a central chemical coding has been revealed
[2, 3, 4].
For example, directly applied in select hypothalamic areas, norepinephrine can produce eating and its precursor (dopamine) can produce the effect after a delay sufficient for synthesis of the active amine; adrenergic blocking agents can reverse or block the effect while acetylcholine in the same area will produce drinking behavior. These effects are contingent upon highly localized concentrations and are not obtained with parenteral or even intraventricular injection. Small changes in the molecular structure of indole or catechol amines can produce differences in psychotomimetic patterns and potency
[5]. With a sleight of the medicinal chemist's hand chlorpromazine becomes the antidepressant imipramine.
Further, drugs have changed our very concept of the way in which familiar behaviors such as sedation are put together. Substituents on the phenothiazine structure produce an alert patient who may show drug-induced restlessness while there is an inhibition of overly activated behavior. Both behaviorally and physiologically the "sedation" or "depression" produced by reserpine is different at different biochemically defined periods
[5, 6] following the administration of the drug; such effects differ from the "sedation" production by phenothiazines
[7].
Apparently, the functional anatomy of familiar behavior patterns is far more complex at both the neural and chemical levels than would be indicated by our inexplicit terminology and while pharmacologists may not always practice this preaching, it is from detailed pharmacological and clinical studies that such distinctions have been made. In brief, these various structure-activity relationships have not only fulfilled earlier promises; they also indicate that we had not sufficiently appreciated the different components, the regulatory and compensatory systems underlying apparently similar behavioral states. Nor did we anticipate the variety of chemically dependent linkages — the associations and dissociations — of which the nervous system is capable.
Limitations of Biochemical Pharmacology
The basic — perhaps preposterous — question posed by psychoactive drugs is how it is that the biochemical change can become behaviorally manifest; in a rigorous sense there is an appalling distance between biochemical mechanisms and the particular substrata for perception and behavior. The question inherently entails a detailed study of intrinsic control systems at a number of different levels from the enzymatic to the psychosocial. Our progress has been of an empirical nature and the rapid proliferation of findings has occurred precisely because we are mapping out the terrain and specifying mechanisms and pathways which, in fact, occur in nature. Yet it is to be doubted that the most extensive encyclopedia of biochemical findings would be sufficient to explain drug-behavior correlations.
If we make a distinction between drug action expressed in biochemical language and drug effect (which is determined by multiple factors) we can agree that a biochemical explanation alone must be of limited value. Every drug acts essentially to facilitate, replace, alter or compete with the special and ubiquitous cellular mechanisms which normally regulate body chemistry. Hence no drug can be confined to effects on behavior. Similarly no behavioral pattern is wholly contingent on drug action, whether we are studying isolated heart muscle or psychological attitude.
The surrounding conditions influence the pattern of effects. Since every drug has more than one action and more than one site of action, since prior state, dosage and route and schedule of drug administration are critical parameters of drug action and effect, the study of each drug requires a careful mapping of significant events. We might recall that simplistic though clear explanations of the effects of psychotropic drugs were available even before the evidence of the past ten years was accumulated and we can continue to expect annual ad hoc explanations from those adroit in the byways of metapharmacology. The fact, however, is that we know far more about peripheral adrenergic nerve and associated responses than we do about the special problems of brain neurochemistry and neurobehavioral effects.
Unless we can couple biochemical sequences to precisely defined sequences of physiological and behavioral responses, unless we sufficiently control and define the effects in which we are interested and more closely link periods of biochemical change with behavioral change, our progress must be restricted.
Advances in Amine Biochemistry
Apart from these caveats, the striking advance of the decade is the fact that at long last we have learned something of the 'detailed biochemistry of those amines
[8] which must underlie fight and flight behaviors. With respect to the catecholamines a number of exotic and endogenous amines have been identified (epinine, octopamine, synephrine). Biochemists possess a range of tools with which to manipulate the various sequences in amine synthesis and inactivation. These tools may be applied easily in vitro and occasionally, with success, in vivo.
Essentially, the task of the body is to convert amino acids from dietary sources into bioactive amines (Figure 1). Amines such as acetylcholine, serotonin, histamine, norepinephrine and its precursor dopamine are highly potent biological substances which, in minute quantities, induce physiologic responses in a number of extraneural tissues, as well as in certain neural systems.
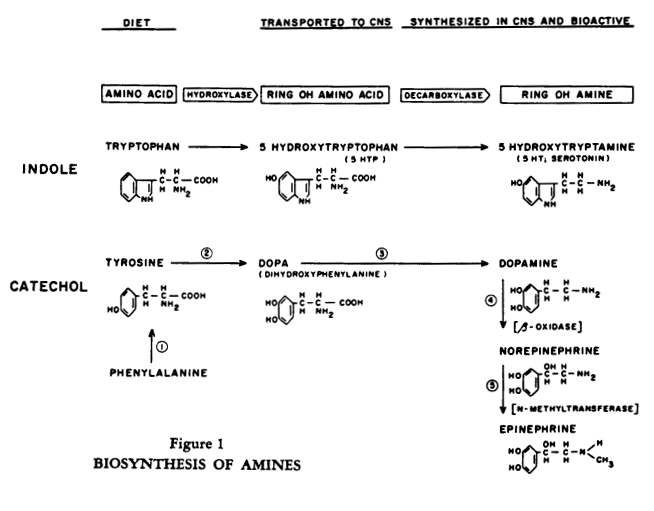
The amine can be synthesized in one location in the periphery and delivered through the circulation to another peripheral tissue; the brain, however, must largely synthesize its own amines from the precursor amino acids and does so in a regionally specific pattern. If we focus on the catecholamines, there are two salient features to their inactivation (Figure 2).
Figure 2:
CATECHOLAMINE METABOLISM
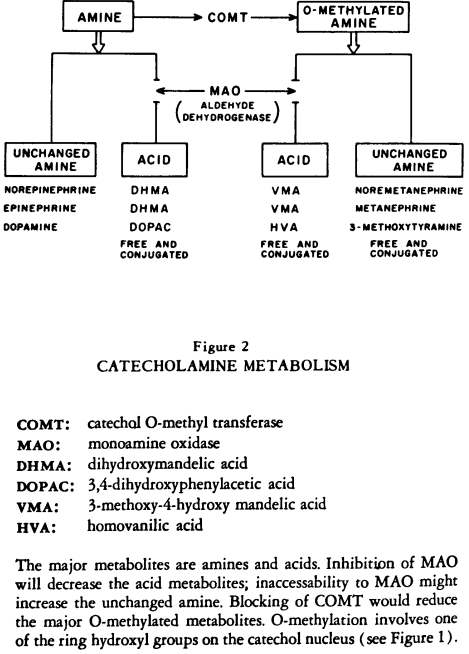
First, the major excretory products are amines and acids (or alcohols). Second, prior to excretion there are two pathways: the amine may be O-methylated or not; therefore the end products of amine metabolism consist either of unchanged amines and acids or of 0-methylated amines and acids (in which one of the ring-hydroxyl groups is 0-methylated).
In a sense, the basic vocabulary is now available and the task of the future is to establish the syntax — the rules regulating the activity of these amines under normal and abnormal conditions and their relationship to energy
[9, 10] and fatty acid metabolism
[11] and to endocrine as well as neural function. We shall later review the significance of some drug effects on the various synthetic and inactivating pathways. Essentially, it is:
- The pattern of amine metabolites.
- Rates of change of tissue levels and the concentrations of amines at various cellular and subcellular sites.
- Manipulation of the various enzymatic steps which provide the biochemical clues with which the pharmacologist can establish relationships to function.
Explanatory Notions and Experimental Definitions
The most generally useful explanatory notion and one which has an ancient and respectable history in pharmacology has held that the behavioral changes following psychotropic drugs are due to excesses and deficiencies of endogenous substances at critical brain receptor sites — or to direct drug effects — or to both. Excesses and deficiencies would be achieved by changes in supply and demand, by synthesis, utilization and destruction; ultimately by binding and release of available substances at the receptor.
These useful notions, while applied to psychotropic drugs a decade ago, nevertheless had to be put into operational terms. The altered balance of amines at a receptor was envisaged but empirical measures were feasible only if large alterations in whole brain levels occurred. Monoamine oxidase (MA) inhibitors produced large increases in amine levels due to decreased destruction of the amines while reserpine caused drops of more than 50 percent. The effect of reserpine was due to a release of amines, presumably from binding sites in storage compartments, and to an impairment of the binding of newly synthesized amine. This observation was of exceptional interest, since binding and release in the absence of fluorimetrically measurable quantities of the drug were linked to a period of altered effects
[13].
Shore, et al.
[12, 13] hypothesized that the now free and unbound (but probably unmeasurable) amine was capable of exerting a prolonged effect. Yet apart from the fact that this could be true for a number of amines following reserpine it was clear that this drug-induced phenomena could not represent the only form of binding and release in nature and that changed levels of this magnitude cannot be the only definition of functional excesses and deficiencies. For example, small elevations in levels of serotonin following the serotonin precursor (5-HTP) produced sedation
[14, 15] and small increases (on the order of 20%) in serotonin levels were observed following LSD (16).
The question also arises as to whether the "normal" variation in levels (changes of approximately 10%) is due to nature or to experimental error. In any event this variation made the measurement of small changes, such as those produced by LSD, precarious, although theoretically all of these small changes could equally represent actual binding and release phenomena.
The problem of technique, therefore, was to find measures more responsive to the concepts and to localize these changed levels in terms of the cellular compartments in which they might be occurring. For example, with the simple technique employed by Giarman and Schanberg
[17, 18] — differential centrifugation — it was shown that the particulate matter of the brain cells contained approximately 70% of the serotonin and this fraction showed the greatest depletion following the full effects of reserpine.
Association of other amines with the granular material of cells both in the periphery
[19, 20] and the brain
[21] had been previously demonstrated. It was found that a variety of psychotropic drugs—whatever their effect on whole brain levels—changed the distribution of amines in the particulate and supernatant cellular fractions which represent a number of different subcellular components
[17, 23, 27].
The notion was that this changed distribution occurred because of the effects of drugs on the normal equilibria of the amines within the cells and that the relative amounts of amines in such fractions following drug treatment were consequential
[23, 24]. In other words, localization was as important as total levels which could represent only the net effect of a number of processes. This work emphasized the basic idea that rates of change and the "traffic" of amines were relevant to observed effects. What would be required were more refined measures to reflect these equilibria and shifts in intracellular concentrations. Thus both biochemical measures of amine levels and turnover rates as well as structural considerations would be necessary to decode the "syntax" governing the operation of the biogenic amines.
Structure-integrated Function
There has, in fact, been a rapid development in techniques in the broad field of biochemical pharmacology as well as in the area of psychotropic drugs: radioisotopes, electron and fluorescence microscopy, density gradient separation and differential centrifugation — all of which are bringing biochemical sequences into closer approximation with the visualizable reality of structure.
The old concept that the cell is little more than a bag chock-full of enzymes has been replaced by a picture of a complex subcellular ultrastructure. This is rapidly becoming biochemically mapped and evacuated in terms of the implication for function of the structural localization of chemical mechanisms. Coupled with our awareness of the regional localization in the brain of bioactive substances, this morphologic emphasis is the hallmark of contemporary work and represents the basis for current interpretations. The key for contemporary biochemical pharmacology is, as Heinrich Waelsch once put it, "structure-integrated function."
Transport of Drugs to Brain Sites
Figure 3 is a diagram of a typical capillary-glial-neuronal assembly and significant subcellular elements (the specialized nerve ending with vesicles, the thickened presynaptic and post-synaptic patches of membrane, mitochondria scattered throughout the channels of endoplasmic reticulum). Unlike most tissues, the brain has little extracellular space and the perivascular glial cells interspersed between blood and neurone may serve as a specialized exchange station analogous to an extracellular space.
Figure 3:CAPILLARY-GLIAL-NEURONAL ASSEMBLY.
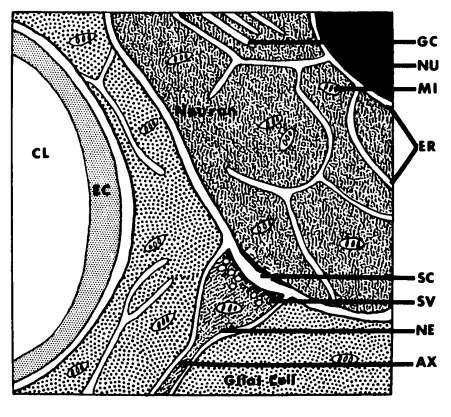
Here, then, is a structural basis for the highly selective — often energy-dependent—exchange of substances between blood and brain conceived of as the "blood-brain barrier." For instance, the selective localization of carbonic anhydrase in glial cells may reflect a role in exchanging carbon dioxide between neuron and blood, and inhibitors of carbonic anhydrase (such as Diamox) may ameliorate certain seizure disorders in childhood. The ultrastructure of capillaries differs in the brain and this allows for differential transport in different regions
[25].
The crucial question in tracing the impact of a drug on the critical bioactive sequences of the body—including the question of lipid solubility and selective transport —is the way in which drugs become accessible to active sites. In general, the amounts of drug required to exert an effect at an active receptor are far less than the amounts required to circumvent the liver and bring drug and receptor into contact. It appears that functionally significant brain regions preferentially concentrate chlorpromazine
[26] (or LSD as recently reported by Snyder, et al.
[27] and confirmed in our laboratories (28)), but extensive knowledge about the concentration and distribution of drugs in the brain is generally lacking.
Similarly (with some interesting exceptions) studies of drug metabolism as reflected in blood and urinary measures have not as yet provided a correlation with pharmacologic effects or with individual differences in therapeutic response. It should be emphasized that blood levels of drugs as well as brain levels of drugs must be important since the chemoreceptors in such structures as the carotid sinus are potent in neurophysiologic regulations
[29] and since certain specialized areas of the brain (such as the area postrema and the intercolumnar tubercles) are highly vascularized and may preferentially concentrate a drug. Such concentration as well as differential uptake and release of a drug at various brain areas must be accounted for in linking the sequence of drug, chemical and behavioral changes.
The Psychotropic Drugs and Energy Metabolism
Drug effects on overall energy metabolism have not generally proved to be differentiating and the overall (as distinct from the regional) oxygen consumption of the brain appears mainly to define the limits for coma or consciousness
[30]. The mitochondrion consists of highly organized enzyme complexes integrated into an enclosed system of membranes — an arrangement permitting the efficient trapping of energy released during the oxidation of glucose. Phenothiazines can inhibit electron transport or uncouple this energy system from the oxidative processes (involving glucose intermediates) through which ADP is phosphorylated to yield/ATP. Yet the phenothiazine tranquilizers do not appear to work by reducing energy supplies stored in ATP; ATP levels, at least in the whole brain, may in fact surprisingly increase after phenothiazine treatment
[31].
Chlorpromazine does, however, quite sensitively inhibit the transition from resting oxidative states to states of high activity in brain slices and in isolated mitochondria
[32, 33]. A more general effect of the phenothiazines is their ability to alter the configuration and permeability of various membranes and thus potentially to influence intercompartmental traffic (34). Speaking far too broadly, one can see that at many levels the effects of phenothiazines are to stabilize an ongoing state; they dampen, for example, the often damaging and overcompensatory physiological responses occurring to certain stressors, by virtue of an action on interneural transmission in certain sites in the brain stem (35, 36).
Neuromodulators and Transmitters
The chief point of convergence in the attempt to link physicochemical phenomena and bodily response has been the synapse where drugs must somehow influence the transfer of information between neurones. A specialized presynaptic structure — the nerve ending—contains "synaptic vesicles" which because of their proximity to the synaptic membrane are thought to be the storage sites allowing for efficient release — or retention of neurohumors; in the periphery, for example, the number of vesicles decreases after stimulation. The presumed transmitter would attach to synaptic membranes and initiate depolarization, or acting as a neuromodulator, the substance would modify the environment in which the transmitter acts
[37].
While norepinephrine is probably the peripheral adrenergic transmitter and while. there is some presumptive evidence for acetylcholine as a transmitter in the brain, it is generally agreed
[38] that we know too little, in fact, to engage in more than guessing games concerning the role of individual brain amines in synaptic transmission or in various neurobehavioral "functions." For purposes of orientation only, we can note that some, though not all
[39], central effects of serotonin tend to be of a sedative nature and possibly some serotonin effects involve cholinergic receptors; excesses of norepinephrine may be crudely associated with certain excitatory states and deficiencies with certain states of exhaustion and sedation.
Levels of acetylcholine tend to correlate with physiologic sleep and excitation. Single-cell recording following electrophoretic administration of chemicals to brain reveal serotonin, norepinephrine and acetylcholine sensitive cells
[42], and with microperfusion techniques, acetylcholine in the brain can be recovered in the effluent following stimulation
[40, 41].
It should, however, be clear that simply because activity can be correlated with application or concentration of a substance in the brain, the substance cannot thereby be directly implicated as a synaptic transmitter or inhibitor. Nevertheless, the imaginative notion put forth by Fatt and Katz
[43] that stored quanta of a transmitter would be released on nerve stimulation has been bolstered by evidence, including the fact that a number of endogenous amines have been found to be stored in vesicles (such as those visualized in nerve endings); these substructures seem to have a capacity for concentrating exogenous amine
[44, 45, 46, 47, 48]. With labeled amines, the uptake of radioactive grains associated with vesicles in sympathetic nerve has been actually visualized (49). With further refinements in technique such findings may yet be more precisely interpreted.
Intact peripheral nerve is required for the uptake and binding of amines and nerve section produces a loss of the vesicular binding sites and supersensitivity of the receptor to injected amine (50). In the peripheral nerve, electrical stimulation (or certain drugs) will produce an efflux of amines
[50, 51, 52].
Similarly, section and degeneration of various tracks in the central nervous system has been shown to produce changes in amine levels and in synthesizing enzymes
[53, 54, 55]. Elucidation of the relationship betWeen neural activity and brain enzymes and substrates, and the effect of central neural activity of hormones
[56] and peptides on binding and release should provide the next major development in biochemical aspects of psychopharmacology.
The Life Cycle of Amines
Figure 4 provides a diagrammatic representation of the life cycle of the brain amines. Psychoactive drugs may act at a number of points in this cycle: the transport of precursors, the synthesis in the cytoplasm of amines, their binding and release in subcellular structures — and their subsequent rebinding — or their enzymatic inactivation. Binding usually means functional inactivation or inaccessability of the amine due to storage in organelles; physically, however, it can occur at the receptor or with soluble proteins in the cytoplasm and such "bound" amines could be functionally associated with activity or not.
Figure 4: MECHANISMS GOVERNING BRAIN AMINE LEVELS
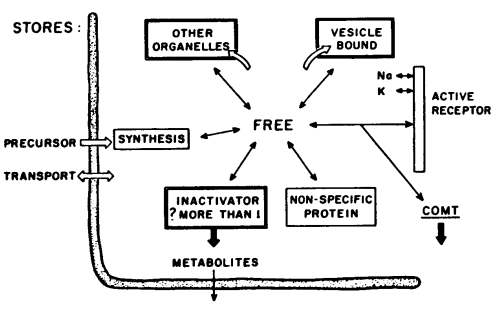
How amines normally get bound or released is not known. The notion that there are pumps, not only at the cell wall for the brain amines but further pumps at the organelles, has yet to be firmly proved. Much work remains to be accomplished — especially in the brain; but when the chemist can localize an enzyme in an organelle (e.g., dopamine beta oxidase in granules
[57], he can then tell whether or not an amine has entered the granule or been shunted elsewhere, and when he can identify hydroxylated metabolites he can deduce, theoretically, where the amine must have been to have undergone the biotransformation.
Some 'sketches' of Amine Traffic
A current view of data is that there are at least two pools of bound amines. The pool conceived to be nearer the receptor represents an easily released and more active pool. Sympathomimetic drugs or amines release this pool and thus indirectly cause hypertensive effects. After repeated dosage the pool is depleted and tachyphylaxis is observed. Catecholamines releasable in this way are thought to be inactivated by catechol-O-methyl transferase (COMT), an enzyme which Axelrod pictures as located in the cytoplasm or extracellular space (58). Drugs such as reserpine induce release also from the deeper lying pool and these amines are shunted to the mitochondria' MAO and inactivated within the cell
[59].
Following reserpine, inactive metabolites may cross the synaptic membrane and, accordingly, there is no hypertensive response. It may also be that reserpine affects the cell wall
[60].
MAO inhibitors sometimes cause hypotension, even though actual levels of norepinephrine are increased. Apparently, these inhibitors alone do not produce a functional excess of norepinephrine. It is possible that the activity of COMT protects the receptor (and opinion varies about an additional effect of MAO inhibitors: inhibition of the release of amines
[58, 61, 62, 68].
In any event, following MAO inhibition the easily-released pool may be still readily triggered by drugs or by amines (such as tyramine found in cheese) and this leads to quite potent hypertensive effects. This may be due to the greater quantities of amine packed in the pools and 'ready' to be released by appropriate chemicals.
Chlorpromazine permits the storage of intracellularly synthesized amine but blocks the uptake of exogenous amines and the reuptake and recirculation of intracellular amines
[58]. It thereby exposes amines more rapidly either to metabolism by COMT or to the receptors
[63]; because chlorpromazine also blocks the receptor, the overall effects are hypotensive.
Imipramine has similar effects but the receptor is not blocked. The effects of imipramine and its active intermediate, desmethyl imipramine (DMI) are markedly enhanced when levels of catechols are changing —as during reserpine-induced release of norepinephrine (64). The mechanism by which imipramine "sensitizes" amine receptors is not clear
[65].
Generally, both categories of antidepressant drugs do not seem to be directly acting, but rather to be sensitizing compounds and their activation effect in clinical depression may depend on the status of endogenous chemical releasers — the effects of which they markedly enhance. The view of antidepressants as sensitizing emphasizes the importance of prior neurobehavioral states upon drug effects. When depression is viewed behaviorally, it is as if a concurrent tension were converted into activity in the presence of these drugs; the functional status of the brain amines in depression becomes, therefore, an intriguing question.
Drugs such as alpha methyldopa (Aldomet) might be called "displacers"
[66]. They and naturally-occurring amino acids compete for the synthesizing mechanism and are themselves converted to amines and stored in granules and released. Hence this leads to the notion of "false transmitters"
[67, 68]. Other amino acidsphenylalanine compete with the serotonin precursor for uptake at the cell wall leading to altered brain amine levels, possibly of significance in phenylketonuria
[69, 70].
Schematic representation of the typical capillary-glial-neuronal arrangement. A glial cell is seen interposed betweetrcapillary and neuron. An axon (AX) with its specialized nerve ending (NE), containing synaptic vesicles (SV) and mitochondria (MI), terminates at the neuron to form a synapse. An extensive system of internal membranes and channels, the endoplasmic reticulum (ER), is present in the neuron and, to a lesser extent, in the glial cells. Mitochondria are scattered throughout intervening areas of cytoplasm (stippled). Other abbreviations: CL: capillary lumen; EC: endothelial cell; GC: golgi complex; NU: nucleus; SC: synaptic cleft.
These sketches of the intricacies governing amine traffic (which were first indicated by differential centrifugation studies) are based largely on investigations from the laboratories of Julius Axelrod and those of Irving Kopin at the NIMH, where the fate of small amounts of highly labeled injected amine in peripheral tissues is investigated. However, the details of such schemes are finally adjudicated; experimental studies show that a gamut of psychotropic drugs — antidepressants, phenothiazines and synpathomimetic amines—could influence the balance of amines at an active receptor.
Such studies have also shown that classical tools in biochemistry can be integrated with structural considerations. In summary, it is now generally accepted that psychotropic drugs influence not only levels of amines, but their movements, sub-cellular localization, rates of turnover and accessibility, not only to receptors and to sites of storage, but to different pathways for enzymatic synthesis and destruction. Drugs may also block or induce enzymes. They may alter physical-chemical properties of membranes and receptor substance, influence ion flux, metabolic processes, or rates of binding and release and thus directly or indirectly influence neural functions.
Psychotomimetic Drugs
A number of active alkaloids isolated from plants and used ritually by natives (and even college students) to induce altered subjective states contain lysergic acid or structures related to biogenic indole and catechol amines (Figure 5).
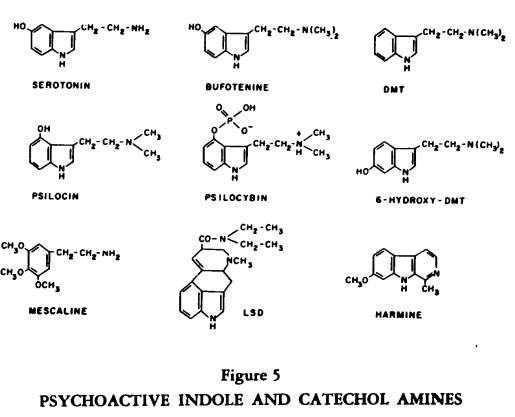
This group of compounds, drugs such as LSD-25, mescaline and psylocin and dimethyltryptamine (DMT) also influence binding and release of endogenous amines (5). Behaviorally, these substances produce a loss of the customary controlling anchors so that the usual boundaries which structure thought and perception become fluid; awareness becomes vivid while control over input is markedly diminished. Dependency either on the surroundings or on prior expectations or on a mystique for structure and support is enhanced.
Psychiatrists can recognize these primary changes as a background state out of which a number of secondary psychological states can ensue, depending on motive, capacity and circumstance. Our terminology reflects this: if symptoms ensue, the term psychotomimetic is employed; and if mystical experience, religious conversion or therapeutic behavior changes are stressed, the term psychedelic or "mind manifesting" has been applied. (And some thirty years ago chemists called the harmine- and harmaline-containing alkaloids, derived from South American cohaba, "telepathine").
The importance of these compounds does not lie essentially in an identity between such drug-induced changes and the operative biochemical pathways in clinical disorders; rather, the drugs offer a tangible grip upon neurochemical sequences related to a peculiar and interesting mental state, and research with such a drug could provide at least one detailed demonstration of how this state can, in fact, be achieved.
The question does arise as to whether mammals can produce endogenous psychotoxins and the enzymatic machinery for this appears to be present. For example, enzymes which can N-methylate indole or catechol amines are known; this means that DMT or bufotenine could be produced in the body. 6-hydroxylation of indoles can occur in the human and this substituent on the N-methyltryptamine structure enhances psychotoxicity in animals. The me thoxylation of catechols leads to a loss of sympathomimetic potency but multiple methylation of phenethylamines leads to psychotoxic effects, as with mescaline. Friedhoff
[71] has evidence that dimethyoxylation of catechols occurs more frequently, perhaps, in schizophrenics than normals. 5-methoxylation of indoles heightens their behavioral effects and this enzymatic reaction occurs normally in the pineal, where N-acetyl serotonin is methoxylated to produce the skin-lightening hormone, melatonin.
Beta carboline structures like harmaline are seen in another pineal hormone, adrenoglomeruloltropin, and Szara has proposed that such structures could be formed by the cyclizing of in-doles, perhaps as a psychoactive intermediate following drugs such as psilocin. Thus, demonstrations of the capacity of the human body to synthesize psychotoxic substances have clearly advanced, although it should be clear that such enzymatic conversions have not been etiologically linked with clinical disorders.
The relative potency of these compounds can be objectively demonstrated in rats working on a simple (FR) schedule in which the rat presses a lever for food
[72]. With these measures LSD is about 150 times as potent as mescaline. The schedule can also demonstrate a period of acute effects of LSD in the rat and the phenomena of tolerance and cross tolerance
[73]. The acute effects following an effective dose (ED90) of LSD in rats begins at five minutes and are over by 30-45 minutes. As the LSD leaves the brain, and the serotonin is bound, these behavioral effects are observed (Figure 6).
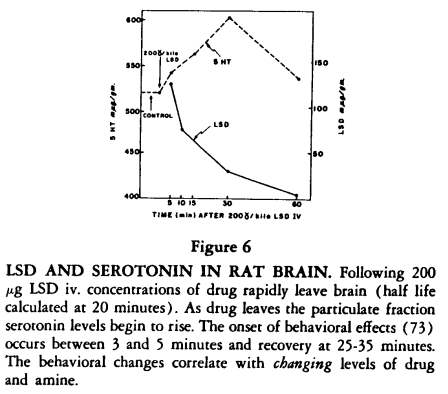
We have recently found that the LSD at five minutes is concentrated in the particulate fraction and as it leaves this fraction and is concentrated in the supernatant, the binding of serotonin begins to occur in the particulate fraction, suggesting that LSD has changed the serotonin receptors there and defining a remarkably close relationship between LSD and the indole receptors
[28].
There are further biochemical links between amine metabolism and LSD. Long after reserpine has been metabolized—or after the effects of a short-acting releaser such as tetrabenazine are over (and amines are only 14% below normal) the effects of LSD in man and rat are enhanced and prolonged; doses of LSD in rats normally ineffective become potent
[74, 75]. This rather surprisingly proved not to be an effect of such drugs on the uptake of LSD in the brain since, in fact, less LSD enters the organ. Rather, there is enhanced duration of the binding of serotonin in the same cellular fraction from which the reserpine and tetrabenazine release amines
[28].
A number of psychotomimetic drugs bind serotonin
[76] and release norepinephrine and such effects have been found with toxic dosage schedules of amphetamine
[77]. Szara finds such effects most striking in the rabbit hypothalamus
[78]. Premedication with certain MAO inhibitors may dampen the effects of LSD
[79]. These and other
[79, 80] studies, then, show that psychotomimetic drugs have an affinity for brain receptors which normally process amines and that the binding of serotonin is somehow related to a factor governing intensity of drug effect.
The presence of the drug in the body is also important. The half-life of LSD in plasma correlates with the duration of intense effects not only in man
[81] but in rat, rabbit and cat, and we have some evidence that during the course of an LSD episode, fluctuations in plasma levels may correlate with periods of intensification of the LSD effect
[28]. So that in spite of low drug levels (a few millimicrograms/cc in plasma) there may be several select receptor sites in the brain which can continue to be stimulated by only a few molecules of drug held in the plasma during the periods of changing behavioral and amine effects.
Very low doses of LSD can produce behavioral effects in man
[82] and animals
[83] and biochemical correlates of these effects have yet to be found. Scopolamine and piperydil glyocollates (Ditran) have no effects on the indole and catecholamines, but induce a 30% drop in levels of brain acetylcholine (84). Therefore, there are several groups of psychotomimetic drugs, both in terms of their behavioral and their chemical effects.
Synthesis and Destruction of Amines
Since the brain must synthesize amine from precursors, precursors have been used not only to test the effects of excessive amine synthesized in situ, but to deduce the "real" effect of the amines. In view of the consequences of compartments for function, this is not an uncomplicated approach, especially if precursors are used after reserpine or MAO inhibitors.
Some drugs used in psychiatry influence various steps in the synthesis of amines (Figure 1); e. g. Antabuse inhibits dopamine beta oxidase. Recently it was reported that norepinephrine synthesis can be blocked at an early step (tyrosine hydroxylase) by alpha methyl tyrosine. This important study by Spector, Sjoerdsma and Udenfriend
[85] suggests that after inhibition we can give precursors later in the sequence of norepinephrine synthesis (e. g., at 3 in Figure 1) and thus explore the role of dopamine in the central nervous system.
Further this inhibition permits the study of utilization — or physiological demand for the amine in various functional conditions — a key to many puzzles concerning the role of catecholamines. Finally the inhibition of synthesis by this method does not alter the binding, release and uptake mechanisms and hence various precursors and sympathomimetic drugs will not have bizarre effects, as observed following drugs which do alter amine regulations (such as MAO inhibitors). The behavioral picture appears to be one of sedation, but a different pattern than that seen with reserpine. The animal appears mainly to be easily fatigued and unable to withstand excitation or temperature extremes.
While the liver 0-methylates much of the circulating norepinephrine (and hence the 0-methylated acid is a predominant urinary metabolite), the effect of drugs on various pathways of amine metabolism in Tissues can nevertheless be monitored (Figure 2). If drugs affect not only enzymes but accessibility of enzyme and substrate—the traffic of amines—we should and do see shifts in metabolic patterns following psychotropic drugs. Thus, following the release of amines by reserpine there is an increase in acid secretion which is followed by a decrease when storage is no longer possible
[86].
Chlorpromazine, which prevents the uptake and re-uptake of the amines — perhaps exposing them to COMT but not MAO — produces a decrease in the 0-methylated acid (86). Following MAO inhibitors there is a marked decrease in the VMA excretion and it has recently been reported that there is a decrease following imipramine (87). These reductions in acid by two antidepressants occur, of course, with different mechanisms: one by enzyme inhibition, the other by a shunting mechanism.
It should be remembered that accurate and fine measurement of even one me tabolite is a formidable methodological problem. What we now require is a gamut of urinary and tissue metabolites to be screened after drugs. When this can be accomplished, it will be possible to deduce, more accurately, tissue events from urinary patterns. Metabolic patterns in the brain, of course, need not be reflected in urinary patterns; nevertheless, there are enzyme defects which are thought to be localized to the brain and in which the urine does produce concordant findings (88). We should soon have an accurate approximation of the contribution of brain metabolism to urinary metabolites. Some estimates are of the order of 20 percent.
Correlations of Metabolism with Stress
Patterns of urinary metabolites may reflect altered amine traffic as well as enzyme changes, but they have not clearly pointed to etiological factors. A welcome and significant change in emphasis has been to correlate changes in the pattern of endocrine or amine metabolites with side effects of drugs (89) or with functional changes occurring in the course of a behavior disorder. If the amines were indeed related to various intensity factors in behavior such a strategy would be more relevant to the behavioral facts than attempts to correlate classical nosology for clinical syndromes with metabolic change.
McDonald, et al.
[86] found increased VMA excretion in chronic schizophrenic patients at the NIMH. This study implies an interesting definition of the stress which chronic schizophrenia may entail. When normal controls were exercising their usual activities they showed increased norepinephrine metabolism, approaching, but not reaching, that of the patients. When the normals were in bed this increase disappeared, but the schizophrenic group still had an increased amine metabolism and in fact a slightly enhanced metabolism.
Coupled with this observation is a study by Kornetsky, et al.
[90] of this population. Postural hypotension was monitored twelve hours following a single dose of chlorpromazine. The patients showed minimal hypotension compared to normals. When the normals exercised for several hours, however, and a postural hypotension test was then applied, the hypotension had for all practical purposes disappeared. In other words, the normal can begin to approximate the schizophrenic's response only with marked exertion; in spite of the apparent relative motor inactivity of the patients, there appears to be a great deal of activity of some kind in being a chronic schizophrenic.
The schizophrenics' active-inactivity or inactive-activity is reflected in an increased metabolism of norepinephrine which is, perhaps, greater even when the patient is resting than when moving, and it is also evident in a relative resistance to the marked hypotensive effects of chlorpromazine. It has been clear for some time that if we are to study effects of stress in a manner relevant to clinical disorders, such as schizophrenia, we shall have to refine our notions of the physiological measures which may be appropriate and recognize and define the nature of the stress which is implied in psychological
[91, 92] and psychotherapeutic studies of the disorder
[93].
In general, the neural and biochemical components of a variety of stressful states have yet to be disentangled and are poorly comprehended by confining attention solely to the adaptation syndrome.
Since drugs act to influence binding and release we would have an elegant rationale for drug therapy, should there be a corresponding pathophysiology of binding and release in the brain, either as a primary or secondary effect of psychosis or depression. There are no apparent direct approaches as yet to this question.
Although we know that chemical states affect neurobehavorial activity, in a basic sense our knowledge of how neurobehavoral activity affects chemical states is minimal
[53,54,55]. It is, nevertheless, of interest that with intense life threatening stress requiring marked exertion — such as the requirement that rats swim for their live — changes in brain amines occur and do so in the absence of the pituitary. These changes correlate with the exhaustion and recovery of the animal — an instance of non-pharmacologically induced change of brain amines
[74].
From our own and other laboratories, there are now reports also of long-term changes seen both in brain catechol amines and acetylcholine in animals raised in various stressful environments
[95,96]. Gillis has observed a shift in the subcellular distribution of norepinephrine in the hearts of young rabbits contingent—simply on the injection of a needle into the peritoneum
[97].
The pattern of brain amine response to acute exhaustion stress and to LSD in rats is strikingly similar. It is tempting to suggest that LSD — without inducing the physical exertion required by swimming — simply initiates a localized brain-stress response chemically.
In other words, there are central components of exhaustion stress which need not be coupled directly to physical activity.
These central components could be activated by chemicals or evoked by surtaxing exertion or extremes of temperature. In this analogy, schizophrenic patients would correspond to LSD activation and the normal subjects to the swimming rats. Local brainstress responses uncoupled from physical activity and reflected in changes of amine distribution or in vegetative responses could be compared to a motor racing with the clutch disengaged.
Finally, we should note a convergence of much more concrete evidence indicating that the brain amines normally may buffer the necessary psychophysiological regulations evoked by stimuli which induce an intense central activation. When we alter the controls normally governing binding and release (by pretreatment with MAO inhibitors or reserpine) and we then induce central activation with drugs or with amino acids (such as methionine or thyptophan) effects are produced which in humans are often described as psychotoxic
[98] or in the case of MAO inhibitors and LSD, the expected psychotoxic effect may be lost. In other words, such pretreatment may have unmasked an underlying "buffer" role of the amines.
If genetic or stress induced impairment of binding and release occurred in clinical disorder, The human would be at a chemical disadvantage in handling life stresses and drugs would be helpful in compensating for these effects.
Leaving fantasy aside, we should note that the work of the future requires further detailed biochemical mapping of brain events and a linking of functional changes with biochemical measures. We still lack sufficient knowledge to have a basis for formal hypotheses which can be accepted or rejected by the critical experiment. For the behavioral and physiological scientist, the challenge is to describe more succinctly the sequences following drug administration, and to define and refine the functional units and behavioral dimensions appropriate for correlation with biochemical change.
Supported by United States Public Health Service grants MHK3-18, 566 and MH03363.
Reprinted, by permission, from Freedman_, Daniel: Aspects of the Biochemical Itharrnacoloky Psychotropic Drugs. lw: Solomon, Philip (Ed.): Psychiatric Drugs. New York: Grune & Stratton,1966
REFERENCES: 1. Elkes, J.: Psychopharmacology: the need for some points of reference. In: A Pharmacologic Approach to the Study of the Mind, ed. by R.M. Featherstone and A. Simon, pp. 26-38: Charles C. Thomas, Springfield, 1959.
2. Miller, N.E., Bailey, C.J., Stevenson, J.A.F : Decreased "hunger" but increased food intake resulting from hypothalamic lesions. Science, 112: 526, 1950.
3. Grossman, S.P.: Direct adrenergic and cholinergic stimulation of hypothalamic mechanisms. Am J. Physiol., 202:872, 1963.
4. Stellar, E.: Drive and motivation. Handbook of Physiology - Neurophysiology Ill, pp, 1501-1527, 1969.
5. Giarman, N.J., Freedman, D.X.: Biochemical aspects of the action of psychotomimetic drugs. Pharmacol. Rev., 17:1, 1965.
6. Giarman, N.J., Freedman, D.X., and Savage, W.L.: Drug-induced changes in the subcellular distribution of serotonin in rat brain with special reference to the action of reserpine. Int. Symposium on Problems of the Brain, Galesburg, 111., March 1-3, 1963. In: Progress in Brain Research, ed. by H. Himwich, p. 72, 1964.
7. Brodie, B.B.: Storage and release of 5-hydroxytryptamine. In: 5-hydroxytryptamine, ed. by G.P. Lewis, pp. 64-83. Pergamon Press, New York, 1958.
8. Axelrod. The formation, metabolism. uptake and release of noradrenaline and adrenaline. In: The Clinical Chemistry of Monoamines, Elsevier Publishing Co., Amsterdam, 1963.
9. Mayer, S., Moran, N.C., Fain, J.: The effect of adrenergic blocking agents on some metabolic actions of catecholamines. J. Pharmacol. and Exp. Titer., 134: 18,1961.
10. Haugaard, N., Hess, M.E.: Actions of autonomic drugs on phosphorylase activity and function. Pharmacol. Rev., 17: 1, 1965.
11. Gordon, R.S., Jr., Cherkes, A.: Production of unesterified fatty acids from isolated rat adipose tissue incubated in vitro. Proc. Soc. Exper. Bio. and Med., 97: 150, 1958.
12. Shore, P.A., Silver, S.L., Brodie, B.B.: Interaction of reserpine, serotonin, and lysergic acid diethylamide in brain. Science, 122: 284, 1955.
13. Shore, P.A.: Review of reserpine. Release of serotonin and catecholamines by drugs. Pharmacol. Rev., 14: 531, 1962.
14. Bogodanski, D.F., Weissbach, H., Udenfriend, S.: Pharmacological studies with the serotonin precursor, 5-hydroxytryptophan, J. Pharmacol. Exp. Titer., 122: 182, 1958.
15. Costa, E., Pschedt, G.R., Van Meter, W.G., Himwich, H.E., Brain concentrations of biogenic amines and EEG patterns of rabbits. J. Pharmacol. Exp. Ther, 130: 1961.
16. Freedman, D.X.: Effects of LSD-25 on brain serotonin. J. Pharmacol. Exp. Ther., 134: 160, 1961
17. Giarman, N.J., Schanberg, S.M.: The intracellular distribution of 5-hydroxytryptamine (HT, Serotonin) in the rat's brain. Biochem. Pharmacol., 1: 301, 1958.
18. Schanberg, S.M., Giarman, N.J.: Drug induced alterations in the sub-cellular distribution of 5-hydroxytryptamine in rat's brain. Biochem. Pharmacol., 11: 187,1962.
19. Blaschko, H., Welch, A.D.: Localization of adrenaline in cytoplasmic particles of the bovine adrenal medulla. Nounyn-Schmiedeberg's Arch. Exp. Path. Pharntak., 219: 17, 1953.
20. Hebb. C.O., Whittaker, V.P.: Intracellular distribution of catecholamines in the choline acetylase. J. Physiol. (Land), 180:1051,1957.
21. Well-Malherbe, H. Bone, A.D.: Intracellular distribution of catecholamines in the brain. Nature (Land), 180:1051,1957.
22. Weil-Malherbe, H., Posner, H.S., Bowles, G.R.: Changes in the concentration and intracellular distribution of brain catcholamines: the effects of reserpine, beta-phcnyl-isopropyl-hydrazine, pyrogallol and 3,4-digydroxy-phenylalanine, alone and in combination]. Pharmacol. Exp. Ther., 132;278,1961.
23. Freedman, D.X., Warman, N.J.: LSD-25 and the status and level of brain serotonin. Ann. N.Y. Acad. Sci., 96: 98, 1962.
24. Giarman, N.J.: Discussion in Symposium on "Effects of Hallucinogenic Drugs in Man," Fed. Proc., 20: 897, 1961.
25. Torack, R.M., Barrnett, R.J.: The fine structural localization of nucleoside phosphotase activity in the blood brain barrier. J. Neuropath. and Exper. Neurol., 23: 46, 1964.
26. Guth, P.S., de Jaramillo, G.A.V.: Phenothiazine distribution in mammalian brain. Fed. Proc., 21: 178, 1962.
27, Reivich, M., Snyder, S.: Regional Localization of LSD in the Brain of the Mon-Key. Fed. Proc., 21: 178, 1962.
28. Freedman, D.X., Croquet, C.A.: Unpublished data.
29. Bonvallet, M., Deli, P., Heilbel, G.: Tonus sympathique et activite electrique corticale. Electroenceph. Clin. Neurophysiol., 6:119,1954.
30. Kety, S.S.: Chemical boundaries of psychopharmacology. in: Man and Civilization: Control of the Mind, ed. by S.M. Farber and R.H.L. Wilson, pp. 79-91. McGraw-Hill, New York. 1961.
31. Weiner, N., Huls, H.N.: Effect of chlorpromazine on levels of adenine nucleotides and creative phosphate of brain. J. Neurochem., 7:180,1961.
32. Mcllwain, H., Greengard, 0.: Excitants and depressants of the central nervous system, on isolated electrically-stimulated cerebral tissues. J. Neurochem., 1: 348, 1957.
33. Aghajanian. G.K.: The effect of chlorpromazine on brain mitochondrial respiration as a function of metabolic state. Biochem. Pharmacol, 12:6, 1963.
34. Gey, K.F., Pletscher, A.: Influence of chlorpromazine and chlorprothixene on the cerebral metabolism of 5-hydroxytryptamine, norepinephrine and dopamine.). Pharmacol. Exp. Ther., 133: 18, 1961.
35. Kollis, J., Bullard, R.W.: The influence of chlorpromazine on physical and chemical mechanisms of temperature regulation in the rat. J. Pharmacol. Exp. The r., 145:373, 1964.
36. KiIlam, E.K.: Drug action on the brain stem reticular formation. Pharmacol Rev.,14: 75, 1962.
37. Giarman, N.J.: Neurohumors in the brain. Yale J. Biol. Med., 32: 73, 1959.
38. Everett, G.M., Wiegand, B.G.: Central amines and behavioral states: a critique and new data. Proceedings of the First International Pharmacological Meeting, Vol. 8, Pergamon Press, 1962.
39. Graff, F.G., Leme, J.G., Rocha e Silva, M.: Role played by catechol and in dolamines in the central actions of reserpine after monoaminooxidase in hibition. int.). Neuropharmacol., 4: 17,1965
40. Gaddum, J.H.: Substances released in nervous activity. Presented: First Int. Pharmacol, Mtg., Stockholm, 1961.
91. Delgado, J.M.R., Rubinstein, L.: Intracerebral release r of neurohumors in unanesthetized monkeys. Arch. Int. Pharmacodyn., 150:530,1964
42. Bloom, F.E., Oliver, A.P., Salmoiraghi, G.C.: The responsiveness of individual hypothalamic neurons to microelectrophoretically administered endogenous amines. Int.). Neuropharmacol., 2: 181, 1963.
43. Fatt, P., Katz, J.: An analysis of the end-plate potential recorded with an intrcellular electrode.j.Physiol., 115:320,1951.
44. Himwich, H.E., Himwich, W.A.: Blogenk amines-Progress in brain research, Vol. 8, Elsevier, Amsterdam, 1964.
45. Michaelson, I.A., Whittaker, V.P.: The subcellular localization of 5-hydroxytryptamine in guinea pig brain.Biochem. Pharmacol., 12: 203,1963.
46. vonEuler, U.S., Lishajko, F.: Effect of adenine nucleotides on catecholamine release and uptake in isolated adrenergic nerve granules. Acta Physiol. Scand. 60: 217, 1964.
47. Gillis, C.N., Giarman, N.J., Freedman, D.X.: Retention of 5-hydroxytryptamine by subcellular fractions of rat brain homogenates. Biochem. Pharmacol., 13: 1457, 1964.
48. Maynert, E.W., Kuriyama, K.: Some observations on nerve-ending particles and synaptic vesicles. Life Science, 3:1067,1964.
49. Wolfe, D.E., Potter, L.T., Richardson, K.C., Axelrod, J.: Localizing tritiated norepinephrine in sympathetic axons by electron microscopic autoradiography Science, 138:441, 1962.
50. Hertting, G., Axelrod, J.: Fate of tritiated noradrenaline at the sympathetic nerve-endings. Nature, 192: 172, 1961.
51. Gertner, S.B., Paasonen, M.K., Giarman, N.J.: Studies concerning the presence of 5-hydroxytryptamine (serotonin) in the perfusate from the superior cervical ganglion.). Pharmacol. Exp. Ther., 127: 268, 1959.
52. Gillis, C.N.: Increased retention of exogenous norepinephrine by cat atria after electrical stimulation of the cardioaccelerator nerves. Biochem. Pharmacol., 12:593,1963.
53. Harvey, J.A., Heller, A., Moore, R.Y.: The effect of unilateral and bilateral medial forebrain bundle lesions on brain serotonin. J. Pharmacol. Exp. Ther., 40:103,1963.
54. Heller, A. Seiden, L.S., Porcher W., Moore, R.Y.: 5-hydroxytryptophan decarboxylase in rat brain: Effect of hypothalamic lesions. Science, 47:887.,1965.
55. Snyder, S.H., Axelrod, J., Wurtman, R.J•, Fisher, J.E.: Control of 5-hydroxytryptophan decarboxylase activity in the rat pineal gland by sympathetic nerves.). PharmacoL Exp. Ther., 147: 3, 1964.
56. Harris, G.W.: Sex hormones, brain development and brain function. Endocrinology, 75: 627, 1964.
57. Potter, L.T., Axelrod, J.: Properties of norepinephrine storage particles of the rat heart.). Pharmacol. Exp.Ther., 142: 299, 1963.
58. Axelrod, J.: The uptake and release of catecholamines and the effect of drugs. In: Progress in Brain Research, ed. by H.E. Himwich and W.A. Himwich, p. 81. Elsevier, Amsterdam, 1964.
59. Kopin, I.J., Gordon, E.K.: Metabolism of norepinopnrine-H2 released by tyramine and reserpinej. Pharmacol, Exp. Titer., 138:241,1962.
60. Meltzer, H.Y.,Barnett, n:, Carlini, G.: Unpublished observations (1963).
61. Pepeu, G., Roberts, M., Schanberg, S.M., Giarman, N.J.: Differential action of iponiazid ("Marsilid". and betaphenylisoprolgydrazine "Catron".) on isolated atria.J)Pharmacot. Exp.Ther., 137:334,1962.
62. Maling, H.M., Highman, B., Spector, S.: Neurologic, neuropathologic and neurochemical effects of prolonged administration of nheavlisonronvlhydrazinc (JB 516), phenylisobutylhydrazine (JB 835), and other monomine oxidase inhibitors. J.Pharmacol. Exp. Ther., 137:223,1962.
63. Gey, K.F., Pletscher, A.: Effects of chlorpromazine on the metabolism of d1- 2-C14-DOPA in the rat. J. Pluoinacol. Exp. Ther., 145: 337, 1964.
64. Sulser, F., Bickel. M.H., Brodie. A.B.: The action of desmethylimipramine in counteracting sedation and cholinergic effects of reserpine -like drugs,). Pharmacol. Exp. Ther., 144-321,1964.
65. Shore, Pa.A., Busfield, D.: The effect of desmethylimipramine on reserpine and insulin-induced release of gastric histamine and adrenal catecholamines. Life Sciences, 3: 361, 1964.
66. Udenfriend, S., Zaltzman-Nirenberg, P.: On the mechanism of the norepinephrine release produced by alpha-methyl-META-tyrosine. J. Pharmacol. Exp. Ther., 138: 194, 1962.
67 Kopin, I.J., Fischer, J.E., Musachio, J.M.. Horst. W.D., Weise, V.K.: False neurochemical transmitters and the mecnanism of sympathetic blockage by monoaximeoxidase inhibitors. J.Pharmacol. Exp. Ther., 147:186.1965.
68. Kopin, I.J., Fischer, J.E., Musachio, J., Horst, W.D.: Biochemistry-evidence for a false neurochemical transmitter as a mechamism for the hypotensive effect of monoamine oxidase inhibitors. Proc. Natl. Acad. of Sciences, 52: 716, 1964.
69. McKean, C.M. Schanberg, S.M. Glarman, N.J.: A mechanism for the indole defect found in experimental phenylketonuria. Science, 137: -7/81/4(4'33/4 -16)
70. Schanberg, S.: A study of the transport of 5-hydroxytryptophan and 5-hydroxytryptamine (serotonin) into brain. J. Pharmacol, Exp. Ther., 139:191, 1963.
71. Friedhoff, A.J. Van Winkle, E.: Conversion of dopamine to 3,4 dimethoxyphenvlacetic acid in schizophrenic patients. Nature, (Land.), 199:127m1963.
72. Appel, J.B., Freedman, D.X.: Unpublished data.
73. Freedman, D.X. Appel, J.B., Hartmen, F.R. Molliver, M.E.: Tollerance to behavioral effects of LSD-25 in rat. J.Pharmacol. Exp. Ther., 143:309,1964.
74. Freedman, D.X.: Studies of LSD-25 and serotonin in the brain. Proc. 3rd Int. World Cong. Psychiat., 1:653, 1961.
75. Appel, J.B., Freedman, D.X.: Chemically-induced alterations in the behavioral effects of LSD-25. Biochem. Pharmacol., /3:861, 1964.
76. Freedman, D.X.: Psychotomimetic drugs and brain biogenic amines. Amer. J. Psychiat., 119:843, 1963.
77. Smith, C.B.: Effects of d-amphetamine upon brain amine content and loco-motor activity of mice.J. Pharmacol. Exp. Ther., 147:96, 1965.
78. Szara, S.: Effect of psychotropic tryptamine derivatives on the regional distribution of serotonin in the brain. Presented at Third Meeting of Collegium International Neuro-Psychopharmacologicum, 1962.
79. Siva Sankar, D.V., Sankar, D.B., Phipps, E., Gold, E.P.: Effect of administration of lysergic acid diethylamide on serotonin-levels in the body. Nature (Loud.), 191:499,1961.
80. Siva Sanker, D.V., Broer, H.H., Cates, N.: Histamine-binding action of lysergic acid diethylamide.Nature, (Lond.), 200: 582, 1963.
81. Aghajanian, G.K., Bind, O.H.L.: Persistence of lysergic acid diethylamide in the plasma of human subjects. Clin. Pharmacol. and Therap., 5:611, 1964.
82. Greiner, T., Burch, N.R., Edelberg, R.:Psychopathology and psychophysiology of minimal LSD-25 dosage. Arch. Neu rol and Psychiat., 79:208,1958.
83. Bradley, P.B., Hance, A.J.: The effects of intraventricular injections of drugs on the electrical activity of the brain of the conscious cat. Electroenceph. Clin. Neurophysiol., 8: 699, 1956.
84. Giarrnan, N.J., Pepeu, G.: The influence of centrally acting cholinolytic drugs on brain acetylcholine levels. Brit.]. Pharmacol., 23: 123, 1964.
85. Spector, S., Sjoerdsma, A., Undenfriend, S.: Blockade of endogenous norepinephrine synthesis by alpha-methyl-tryosine, an inhibitor of tryosine hydroxylase J. Pharmacol. Exp. Titer., 147: 86, 1965.
86. McDonald, R.K., Weise, V.K.: The excretion of 3-methoxy-4-hydroxymandelic acid in normal and in chronic schizophrenic male subjects. Psychiat. Res., 1: 173, 1962.
87. Schildkraut, J.J., Klerrnan G.L., Hammond, R., Friend, D.G.: Excretion of 3-methoxy-4-hydroxymandelic acid (VMA) in depressed patients treated with antidepressant drugs. J. Psychiatric Res., 2: 1964. •
88. Levin, B., Mackay, H.M.M., Oberholzer, V.G.: Argininosuccinic aciduria: an inborn error of amino acid metabolism.Arch. Dis. Child., 36:622, 1961.
89. Bozzi, R., Bruno, A., Allegranza, A.: Urinary metabolites of some monoamines and clinical effects under reserpine and chlorpromazine. Brit. J. Psychiat., 111: 176, 1965.
90. Vates, T., Korenetsky, C.: A comparison of some physiological changes in normal and schizophrenic subjects twelve hours after chlorpromazine administration. Abstract -Fall meeting, American Society for Pharmacology and Experimental Therapeutics, p. 35, 1958.
91. Shakow, D.: Psychological deficit in schizophrenia. Behavioral Science, 8: 275, 1963.
92. Callaway, E., III, Jones, R.T., Layne, R.S.: Evoked responses and segmental set of schizophrenia. Arch. Can. Psychiat., 12: 83, 1965.
93. Pious, W.L.: Hypothesis about the nature of schizophrenic behavior. In Psychotherapy of the Psychoses, ed. by A. Burton. Basic Books, New York, 1961.
94. Barchas, J.D., Freedman, D.X.: Brain amines: response to physiological stress. Biochem. Pharmacol., / 2: 1232,1963.
95. Rosenzweig, M.R., Krech, D., Bennett, E.L., Diamond, M.C.: J. Comp. Physiol. Psychol., 55: 429, 1962.
96. Geller, E., Yuwiler, A., Zolman, J.: Effects of environmental complexity and training of constituents of brain and liver. Abstract, International Neurochemical Conference, Oxford, Eng., July, 1965. (in press).
97. Gillis, C.N.: Unpublished data.
98. Pollin, W., Carrion, P.V., Jr., Kety, S.S.: Effects of amino-acid feeding in schizophrenic patients treated with iproniazid. Science, 133: 104, 1961.
Download Our Free Psychedelic Healing Books



